
第一作者:刘露
通讯作者:胡敬平 教授
通讯单位:华中科技大学
论文引用:Chemosphere, 2023, 331, 138783
论文DOI:10.1016/j.chemosphere.2023.138783

图文摘要
成果简介
近日,华中科技大学环境学院FunMat课题组胡敬平教授团队在《Chemosphere》上发表了题为“Peroxymonosulfate activated by natural porphyrin derivatives for rapid degradation of organic pollutants via singlet oxygen and high-valent iron-oxo species”的研究论文(Chemosphere, 2023, 331, 138783,DOI:10.1016/j.chemosphere.2023.138783)。研究以天然卟啉衍生物叶绿素铁钠盐(SFC)作为催化剂,结合过硫酸氢盐(PMS)用于有机污染物的快速去除。研究表明,体系可在pH为3~11的范围内对双酚A达到几乎100%的去除率,对常见的高浓度无机阴离子具有强耐受性。其中对双酚A的去除起主要贡献的活性物种是单线态氧(1O2)和高价铁氧物种(P+•Fe=O),通过密度泛函理论(DFT)和从头算分子动力学(AIMD)揭示了P+•Fe=O和1O2的产生机制。
摘要
本论文系统地研究了过一硫酸钠(PMS)与叶绿素铁钠(SFC)(一种从富含叶绿素的物质中提取的天然卟啉衍生物)的活化作用,以快速降解双酚 A(BPA)。在初始双酚A 浓度为20 mg/L、pH = 3的条件下,SFC/PMS能够在前10分钟内降解 97.5% 的双酚A,而传统的 Fe2+/PMS 在相同条件下只能去除22.6%的双酚 A。它在 3-11 的宽pH值范围内表现出突出的适应性,能完全降解污染物。此外,还观察到它对高浓度无机阴离子(100 mM)具有显著的耐受性,其中(双)碳酸盐甚至可以加速降解。非自由基氧化物种,包括高价铁氧卟啉物种和1O2被确定为主要物种。特别是通过实验和理论方法证明了1O2在反应中的生成和参与,这与之前的研究结果有很大不同。密度泛函理论(DFT)计算和从头算分子动力学(AIMD)模拟揭示了特定的活化机制。研究结果揭示了铁(III)卟啉对PMS的有效活化,所提出的天然卟啉衍生物将成为废水处理中有效去除复杂水介质中难降解污染物的理想候选物质。
亮点
·在较宽的 pH 值和各种阴离子存在下,90%以上的双酚A在初始10分钟内被有效去除
·首次报道了导致生成 1O2的铁复合物的类过氧化氢酶活性
·令人信服地证实了 P•+FeIV=O 双键在双酚 A 降解过程中的参与
引言
近几十年来,基于过硫酸盐的高级氧化工艺因其产生的强氧化性或高选择性物种而受到越来越多的关注。过硫酸盐可以通过施加外部能量来活化或者化学方法,其中铁基材料由于环境友好性和生物相容性备受青睐。但传统技术通常受限于pH范围和水体共存阴离子的影响。与基于自由基的氧化体系相比,非自由基氧化体系通常表现出更为明显的优势,即良好的pH适应性和对无机阴离子的不敏感性。包括铁卟啉和酞菁等类酶铁配合物被用于活化过氧化物时可通过产生非自由基物种(高价铁氧)去除污染物,并且表现出较宽的pH适用范围和较强的无机离子耐受性。然而,此类催化剂在实际应用中通常受合成成本高和产率低的限制。而从天然物质如大蒜叶和蚕砂中提取得到的叶绿素铁钠盐(SFC)是一种很有前途的替代物质。以PMS为铁卟啉氧化剂时,高价铁氧物种和SO4•−在是主要的活性物种。反应过程中形成的关键中间体即铁(III)-过氧硫酸盐物种(PFeIII–OOSO3, P表示卟啉配体)中过氧键的裂解方式会影响活性物种种类,异裂导致P•+FeIV=O的生成,而均裂产生PFeIV=O和SO4•−。类过氧化物酶和类过氧化氢酶活性是类酶铁配合物的两种反应类型,它们在铁配合物的反应中经常相互竞争。前者将2个氧化当量从氢过氧化物转移到还原底物上,而后者促进氢过氧化物歧化为O2。因此,类过氧化氢酶活性通常被认为是一种不受欢迎的特性。然而,在本研究中,在氢过氧化物的歧化过程中却观察到1O2的产生。而到目前为止,还没有关于类酶铁络合物催化循环中1O2生成的报道。
图文导读
催化降解BPA的性能
以双酚A为目标污染物对投加的催化剂和氧化剂的浓度进行优化。通过各反应体系的对比体现了SFC/PMS体系优越的降解性能。在含酚工业废水中双酚A的高效去除则表明了体系在实际应用中的可靠性。
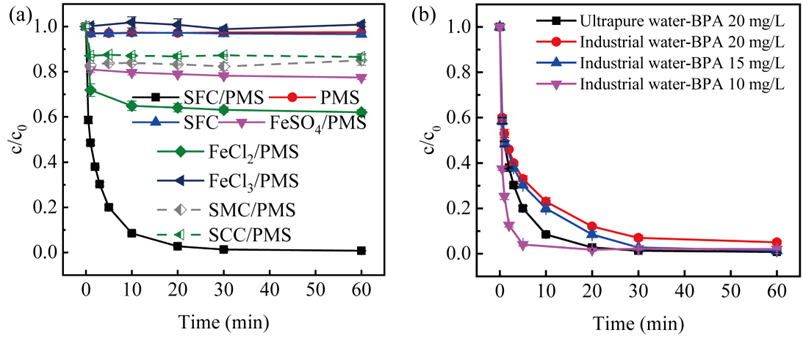
图1. SFC/PMS体系降解BPA的性能
活性物种识别
对体系中的活性物种进行了鉴定。以甲醇(MeOH)、异丙醇(IPA)、叔丁醇(TBA)、叠氮化钠(NaN3)、三氯乙烷(CHCl3)为淬灭剂,发现体系中的活性物种是1O2,而非SO4•−, •OH和O2•−。电子顺磁共振光谱(EPR)检测出DMPOX信号,该峰由DMPO被强氧化性物质氧化产生,根据文献报道,该物质很有可能是高价铁氧物种。采用苯甲基亚砜(PMSO)及2,2-联氮双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(ABTS)探针和环己烯环氧化等进一步证实高价铁氧物种的产生。以9,10-二苯基蒽(DPA)探针进一步证实1O2的产生。

图2. SFC/PMS体系活性物种的识别及高价铁氧物种的鉴定
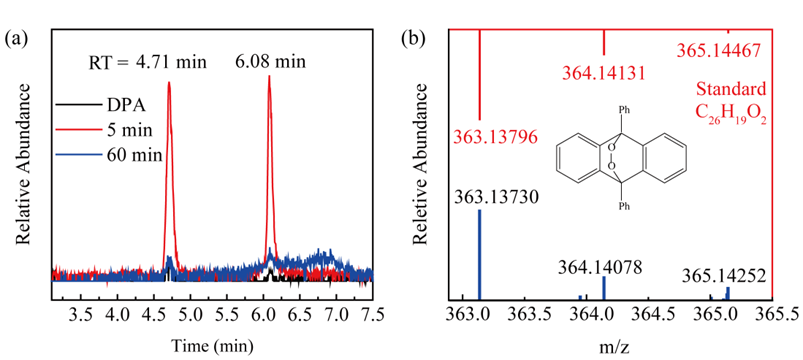
图3. SFC/PMS体系中1O2的鉴定
活性物种产生机制
通过理论计算,包括DFT计算和AIMD模拟,研究了SFC/PMS体系中高价铁氧物种和1O2的产生机制,即PMS首先与SFC中的Fe进行轴向配位,受到卟啉环共轭电子云的影响后,过氧键逐渐拉伸断裂。经过对中间体各片段原子电荷(ADCH)、Mayer键级、自旋布居、氧化态的分析,证明过氧键发生异裂的同时Fe和P发生单电子氧化生成高价铁氢氧卟啉阳离子自由基(P+•Fe-OH)和SO42−,H以质子形式脱离最终形成高价铁氧卟啉阳离子自由基(P+•Fe=O)。由于铁配合物中存在的类过氧化氢酶活性,P+•Fe=O可能和PMS继续反应生成1O2。

图4. SFC/PMS反应生成高价铁氧物种(P+•Fe=O)路径s的吉布斯自由能变化
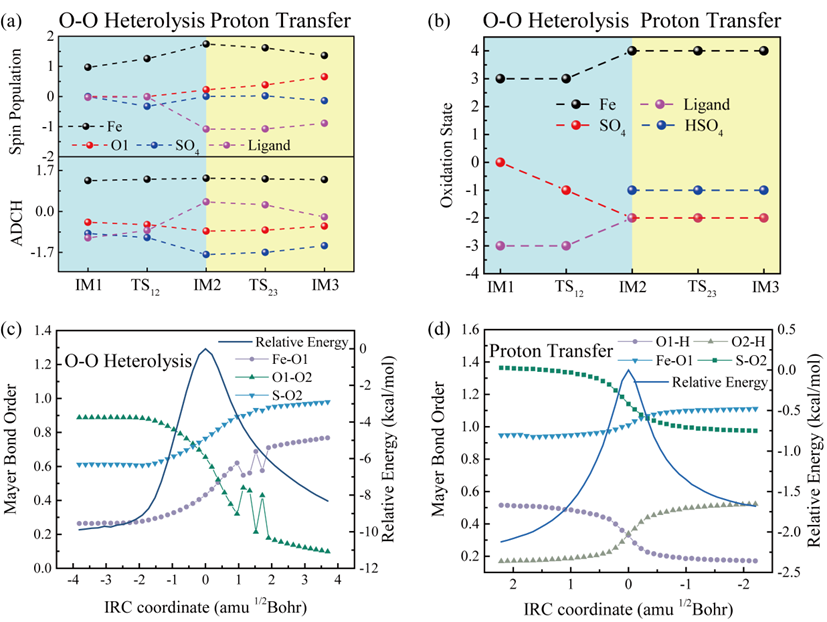
图5. SFC/PMS反应生成P+•Fe=O的电子转移机制
视频1:AIMD模拟P+•Fe=O和PMS生成1O2的反应
影响因素
对体系中催化剂催化性能的影响因素进行探究,发现SFC/PMS能在pH为3~11的宽范围内实现几乎100%的双酚A降解。高浓度无机离子如Cl−、SO42−、HCO3−、NO3−、CO32−对双酚A的降解效率几乎没有影响。

图6. pH和无机阴离子对SFC/PMS体系降解BPA的影响
毒性测试
SFC/PMS对双酚A的矿化率约为30%,以费氏弧菌生物发光抑制实验和微生物燃料电池评估SFC/PMS处理的双酚A降解产物的急性毒性。结果表明,经SFC/PMS体系处理后的双酚A溶液几乎没有生态毒性。
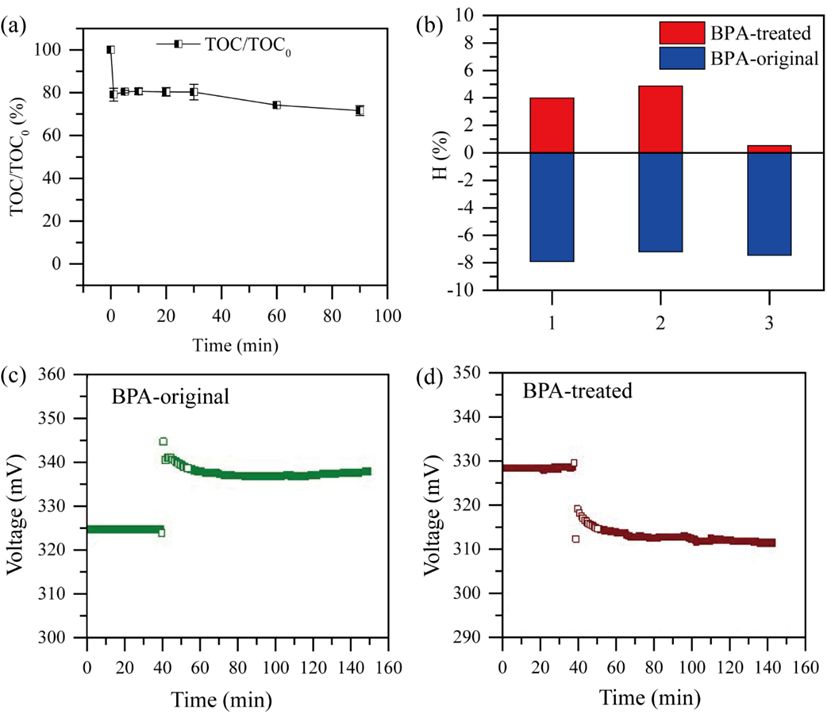
图7. BPA降解矿化率及其毒性评估
小结
本研究利用天然卟啉衍生物叶绿素铁钠盐活化PMS,该体系能够快速高效地去除双酚A,适用pH范围广且对高浓度无机阴离子表现出很强的抗干扰能力。其氧化机制为高价铁氧卟啉阳离子自由基和单线态氧主导的非自由基氧化。通过实验表征、DFT计算和AIMD模拟揭示了活性物种的产生机制,即PMS的过氧键在SFC的活化下首先发生异裂产生P•+Fe-OH和SO42−,H以质子形式脱离最终形成P•+Fe=O。1O2由铁配合物中存在的类过氧化氢酶活性和PMS继续反应生成。这与具有自由基攻击途径(主要是 SO4•− 和 •OH)的传统 PS-AOP 有很大不同,也与所报道的铁络合物/过氧化物系统明显不同。这项研究指出了一种新的过硫酸盐活化途径,为理解过硫酸盐高级氧化体系独特化学性质提供了新的视角。1O2的产生弥补了传统铁配合物/过氧化物体系耗费过量氧化剂的缺陷,而所提出的天然卟啉衍生物/PMS 系统可能是在废水处理中有效消减难降解有机污染物的理想候选物质。
作者简介

刘露,博士研究生,华中科技大学环境学院,研究方向为基于过/亚硫酸盐的高级氧化技术。

王安琪,硕士毕业于华中科技大学环境学院,研究方向为基于过硫酸盐的高级氧化技术。

胡敬平,教授、博导,华科大环境学院实验中心主任,入选国家人才项目(青年项目)。2008年博士毕业于英国牛津大学,博士毕业后先后在英国诺 丁汉大学和牛津大学从事博士后研究工作,2011年当选牛津大学拉姆齐研究学者(Ramsay Fellow)。聚焦环境科学与工程领域,开展固废资源化、环境仿生催化、环境大数据的研究。主持英国The Ramsay Memorial Fellowships Trust项目、国家自然科学基金面上项目与青年项目、湖北省自然科学重点基金、国家重点研发计划固废专项的课题,承担科技部青年973项目和国家自然科学基金创新群体项目,在Adv. Mat.、Angew. Chem.、Adv. Func. Mat.等期刊发表论文190余篇,4篇论文入选ESI高被引论文,H因子45,其中近五年第一作者及通讯作者论文32篇;单篇最高他引220余次,总他引超过6500次,H因子45(Jingping Hu (0000-0001-9984-6636) (orcid.org)),授权发明专利10余项(含3项国际发明专利)、软件著作权2项。担任国际期刊Energy & Environmental Materials副主编、能源环境保护期刊青年编委、巴塞尔公约亚太区域中心化学品和废物环境管理智库专家、湖北省资源综合利用协会专家委员会委员。
文献链接:
论文引用:Chemosphere, 2023, 331, 138783
论文全文链接:https://doi.org/10.1016/j.chemosphere.2023.138783
FunMat课题组微信公共号:

FunMat课题组主页:http://funmat.ese.hust.edu.cn/
 人才招聘
人才招聘
 环境学院公众号
环境学院公众号
 环境学院视频号
环境学院视频号